


鱼类病毒性神经坏死病是由神经坏死病毒(Nervous necrosis virus,NNV)引起的一种全球范围的鱼类流行性传染病。NNV为海水鱼中较常见、危害严重的传染病之一,至今已报告的受害鱼类有鳗鲡目(Anguilliformes)、鲈形目(Perciformes)、鲽形目(Pleuronectiformes)、鲀形目(Tetraodontiformes)、鳕形目(Gadiformes)中的40余种[5-6],其中被感染的种类集中在石斑鱼、鲈鱼,在中国受影响最大的是赤点石斑鱼和斜带石斑鱼。对石斑鱼而言,神经坏死病的高发期为鱼类的幼鱼阶段,孵化后的1~3周为发病高峰期,严重时发病率可达100%[7],幼鱼成活率低于10%。神经坏死病的病症通常表现为鱼苗在水中以螺旋状旋转为主的异常游动,伴随着食欲减退,静止时腹部向上。组织学检测发现细胞空泡化主要集中发生于中枢神经系统细胞以及视网膜[8]。受感染的幼鱼绝大多数在短期内死亡,因此常常导致人工育苗失败,或成活率极低。近年来,随着养殖密度的不断提高和受感染鱼类种类增加,其危害程度愈发严重。
选育抗病品种是解决养殖鱼类病害问题的一个有效途径[9-10]。但是传统的选育方法进展慢,效果较差。2001年Meuwissen T H E等[11]提出了基因组选择(Genomic selection,GS)的方法,该方法具有不需构建家系、育种值估计的准确性高、育种效率高,并可以有效控制近交等多方面优点。如今,随着高通量DNA测序成本不断降低,已有越来越多的水产动物育种开始使用基因组选择的方法[12⇓⇓⇓⇓⇓-18]。目前已经有一些研究者将基因组选择应用于鱼类抗病育种研究,如Tsai H Y等[19]报道了对大西洋鲑抗海虱、Liu Y等[20]报道了对牙鲆抗爱德华氏菌的基因组选择研究。Palaiokostas C等[21]对欧洲鲈鱼(Dicentrarchus labrax)的神经坏死病毒病抗性进行评估,发现使用基因组选择的方法与比系谱选育预测能力增加13%。福建省水产研究所石斑鱼研究团队已经完成了赤点石斑鱼全基因组测序组装[22],华南农业大学Yang M等[23]通过对100尾赤点石斑鱼神经坏死病毒(Red-spotted grouper nervous necrosis virus,RGNNV)易感与抗性石斑鱼进行全基因组关联分析,找到了一些抗病相关的单核酸多态性(Single nucleotide polymorphisms,SNPs)位点;但迄今还没有见到对赤点石斑鱼抗神经坏死病基因组选择研究的报道。本研究对460尾赤点石斑鱼神经坏死病易感(染病死亡,230尾)和抗性(最终健康存活,230尾)鱼苗,通过基因组重测序获得高密度的SNPs集,进行抗病遗传力评估和基因组选择预测,以期为后续的抗病育种实践提供必要的理论参考。
1 材料与方法
1.1 样本的采集与鉴定
实验材料采集于厦门刘五店,该育苗场在2019年赤点石斑鱼育苗生产中遭遇了神经坏死病,分别在发病时期采集已染病的濒死个体作为易感组(共230尾),渡过发病期后,采集同样数量存活的健康个体作为抗病组。对460尾鱼苗采集全鱼,保存于95%乙醇中,用于DNA提取。发病鱼苗病症除了根据其病状判断外,还提取30尾病鱼头部组织DNA制成10个混合样品,用RGNNV特异性检测引物(正向引物:5’-CACCGCTTTGCAATCACAATG-3’,反向引物:5’-GTCATCAACGATACGCACTAGG-3’)进行了PCR扩增验证(结果均为阳性)。
1.2 基因分型和质量控制
使用南京诺唯赞生物科技股份有限公司的快速组织基因组DNA试剂盒提取鳍条组织基因组DNA,进行质检和建库后,在Illumina NovaSeq 6000平台(Illumina,USA)进行WGS测序。其中450尾的目标测序深度为4×,另随机挑选10尾测序深度为20×,用于提供高质量的SNPs变异参考。首先使用fastQC(https://www.bioinformatics.babraham.ac.uk/projects/fastqc)对测序数据进行质量检测,后使用fastp[24]对测序数据进行过滤。最后使用MultiQC[25]对最终的质控结果进行汇总检查。
通过BWA-MEM[26]将clean reads 比对到赤点石斑鱼基因组[22]上,将产生的文件利用Samtools 进行排序并转化为bam文件格式。之后使用sambamba[27]对bam中构建文库时的PCR重复进行标记,对标记重复后的bam文件使用GATK[28]中的“HaplotypeCaller”的模块进行单个样本变异的检测,再通过“CombineGVCFs”将单个的变异集整合为群体的VCF(Variant call format)文件。使用“SelectVariants”“VariantFiltration”模块进行硬过滤以及双等位基因的提取,并使用BCFtools[29]对缺失率大于20%的变异进行过滤,然后对缺失的基因型使用Beagle[30]进行填充。最后通过PLINK[31]对VCF进行过滤以及格式转换,过滤标准为:1)次等位基因频率MAF>0.05;2)HWE<1e-6。最终获得460个样本的高质量SNPs数据用于后续分析。
1.3 群体结构分析
利用PLINK的PCA模块进行主成分分析(Principal component analysis,PCA)。根据PCA结果所占比重前2个主成分PC1~PC2的数据,通过在线绘图工具bioinformatics(http://www.bioinformatics.com.cn)绘制PCA图。
1.4 遗传参数估计
使用GCTA[32]和R语言的EMMREML包(https://CRAN.R-project.org/package=EMMREML)对遗传参数进行估计以及后续基因组育种值(Genomic estimated breeding value,GEBV)的计算。基因组最佳线性无偏预测(Genomic best linear unbiased prediction,GBLUP)模型公式为:
其中y代表的是性状,Z是根据SNPs效应而设计的矩阵(基因型“AA”、“Aa”和“aa”分别对应着SNPs基因型中的“0”、“1”和“2”),β是固定效应(地点效应),混合线性模型也可以展开表示为:
其中λ为惩罚因子,公式为:λ=
1.5 交叉验证
交叉验证用于测试预测的准确度,本研究采用5折的交叉验证,将460尾个体随机分为5组(参考群体与测试群体比例为4∶1),使用GBLUP对460个体的GEBV进行计算,测试群体的表型设置为空,通过参考群体的表型对测试群体的表型进行预测,比较两个值之间的相关系数来衡量模型的预测能力。重复以上步骤5次,使得每一组均有一次机会作为候选群体。交叉验证的重复次数为100次。
1.6 不同标记密度对预测能力的影响
为考察不同SNPs数量GEBV的预测力,除了使用全部的SNPs(6 132 865个SNPs,6 132 k)外,还设计了13个不同个数的标记子集,分别为0.5、1、3、5、10、30、50、100、250、500、1 000、2 500 k。为了降低抽样的误差,使用GATK SelectVariants包对每个数量的标记集都进行50次的随机抽样后,对每次抽样的结果都进行100次的5折交叉验证。
1.7 不同覆盖度标准对预测的影响
根据本研究使用原始的测序深度,对每个个体在每个SNPs上的覆盖度设置了6个过滤标准,即DP3、DP4、DP5、DP6、DP8和DP10,分别对应于3×、4×、5×、6×、8×与10×的reads覆盖度。使用过滤后的VCF文件,通过BCFtools对于每个SNPs位点大于80%的个体的基因型覆盖度大于过滤标准,就保留该位点。过滤后使用BEAGLE对VCF文件进行填充,后对每个过滤标准产生的SNPs标记集进行100次5折交叉验证。
2 结果
2.1 标记分型及群体结构分析
本研究对460尾赤点石斑鱼苗进行基因组重测序,共获得2.67 Tb clean data数据挖掘SNPs,通过哈代-温伯格平衡(HWE)>10-6以及次要等位基因频率(MAF)>5%经过质控后,最终用于分析的SNPs共有5 412 683个(未进行覆盖度过滤),平均标记密度约为200.1 bp/SNPs。对460个样品进行主成分分析,提取前2个主成分(Principal components,PCs)的结果(图1),可以看出群体存在明显的分层现象,分成了2个小的聚类群,但易感(Case)和抗性(Control)个体在两个亚群中均匀分布。
图1
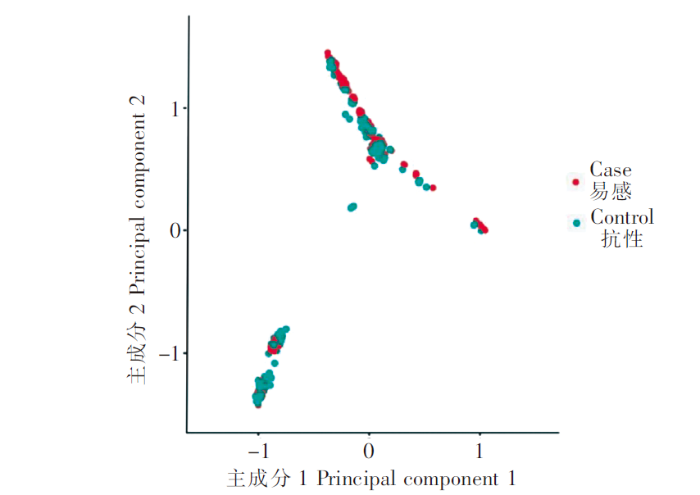
图1
460尾个体的群体遗传结构的主成分分析
注:Case组为易感组;Control组为抗性组。
Fig.1
Principal component analysis of population genetic structure of 460 individuals
Notes:Case group was the susceptible group;Control group was the resistant group.
2.2 遗传参数估计及基因组选择预测
利用全部460尾鱼苗的表型(230尾易感、230尾抗性)和全部SNPs位点的基因型数据计算抗神经坏死病性状的遗传力,使用R包EMMREML的结果为0.566 2,GCTA的AI-REML经过1 000迭代的结果为0.566 6,两种方法估算结果相近。对460尾赤点石斑鱼进行抗神经坏死病基因组选择的预测力分析,80%个体作参考群,20%个体作验证群,分别进行了100次随机抽样的交叉验证,平均的基因组预测准确度为(0.359±0.019)。
2.3 不同SNPs数量的基因组选择预测力
图2
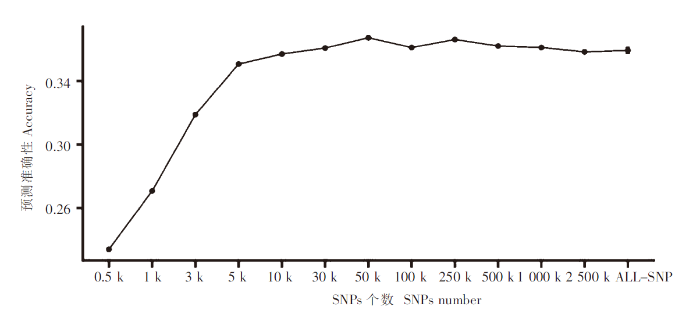
图2
不同标记密度下对赤点石斑鱼抗病性状的预测准确度
Fig.2
Accuracy of prediction model under different marker densities for disease resistance traits of Epinephelus akaara
2.4 不同覆盖度标准对预测的影响
表1 不同过滤标准剩余的SNPs数
Tab.1
| 过滤标准 Filter standards | 填充后 After filling | 过滤MAF后 After MAF filter |
|---|---|---|
| ALL_SNPs | 20 795 120 | 5 412 683 |
| DP3 | 418 632 | 94 992 |
| DP4 | 105 269 | 11 291 |
| DP5 | 56 794 | 5 982 |
| DP6 | 34 427 | 3 695 |
| DP8 | 16 526 | 1 817 |
| DP10 | 7 993 | 830 |
图3
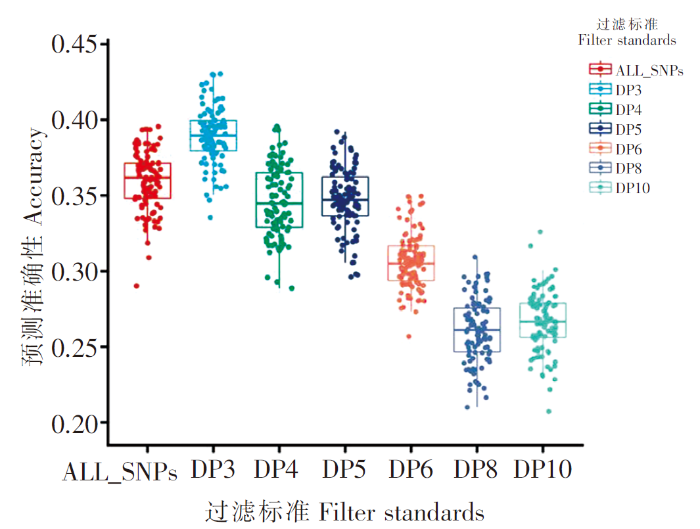
图3
不同覆盖度标准基因组估计的准确性
Fig.3
Accuracy of genome selection estimation for different coverage filter standards
3 讨论
基因组选择的首要工作建立参考群,利用参考群估算各个分子标记位点对性状表型的效应。本文建立了460尾石斑鱼的参考群,主成分分析显示群体聚集为2个不同的亚群,表明群体间存在较分化的亲缘关系。赤点石斑鱼人工育苗中亲本雌雄配比一般达到(20~30)∶1,使用的雄性亲本数量非常有限,因此同一批、尤其是同一池的受精卵可能来自少数雄性亲本。本研究发现鱼苗亲缘关系分化成2个亚群,推测可能采样的鱼苗来自于2个或几个雄性亲本及为数不多的母本。
此外,基因组选择准确性还受基因组的标记密度的影响[36]。本文通过随机抽样研究了不同标记密度对育种值预测准确性的影响,当使用5 k个标记时,即可使预测准确性接近利用全基因组SNPs的水平,其后随着标记个数增加,预测准确性变化趋于平缓;当标记个数为50 k时,预测准确性与利用全基因组所有SNPs的预测准确性几乎一致,这与Tsai H Y等[19]对大西洋鲑抗海虱性状基因组预测的研究结果非常相似,提示如果设计赤点石斑鱼育种芯片,50 k的标记密度可满足育种要求。尽管如此,利用全基因组SNPs标记有利于挖掘具有较大效应的分子标记及因果突变,将这些大效应分子标记和因果突变嵌入预测模型,建立基于主效-微效多基因效应相结合的预测模型,能进一步提高育种值预测准确性。利用同一批数据,笔者在基因组上已经发现了两个效应值极强的GWAS信号(结果未列出),计划在信号内部挖掘可靠的分子标记或因果突变,并将其作为协变量加入GBLUP模型中,通过主效标记/因果突变进一步吸收残余误差的方式进一步提高预测准确性。对测序数据进行覆盖度过滤能够提高对挖掘的标记位点基因分型的准确性,从而在一定程度上提高育种值估算的准确性(图3)。但是如表1和图3所示,在每个个体测序量不变的情况下,随着覆盖度过滤标准提高,可保留用于分析的标记数量急剧减少,当DP≥4时,尽管标记分型的准确度会明显提升,但是由于保留的标记数量减少,育种值预测准确性已低于不进行覆盖度过滤。据Zhang W等[37]对大黄鱼研究的结果,采用全基因组重测序,由于可挖掘到的标记数多,即使测序覆盖度低至0.5×,而且不进行覆盖度过滤,也能获得与8×覆盖度测序基本一致的基因组选择效果,这将大大降低候选亲本标记分型费用。因此,可以预期,随着高通量DNA测序价格进一步降低,基因组重测序将越来越多被用于基因组选择,也将成为石斑鱼全基因组选择育种的主要工具。
参考文献
海水养殖鱼类病毒性神经坏死病防控技术研究进展
[J].
神经坏死病毒(Nervous necrosis virus, NNV)是一种能导致海水鱼脑、中枢神经及视网膜等神经系统坏死的病毒,该病毒引发的鱼类病毒性神经坏死病主要发生在稚鱼和幼鱼期,较强的致病力及高致死率给海水鱼养殖业带来了沉重的打击,成为海水鱼类养殖产业可持续发展最重要的限制因素之一。文章主要综述了NNV的基本特征、诊断技术、病毒传播、疫苗免疫学等研究进展,介绍了NNV的防控技术及策略,为海水养殖鱼类NNV的防控工作提供参考。
斜带石斑鱼(Epinephelus coioides)病毒性神经坏死病毒的纯化分析及检测方法建立
[J].根据GenBank中已有的鱼类病毒性神经坏死病毒(NNV) RNA2基因序列,设计引物,从福建厦门具有典型NNV发病症状的斜带石斑鱼中克隆了RNA2基因的全长序列,并将序列提交到GenBank获得登录号为MF510920,命名为XMNNV。系统进化树分析结果表明XMNNV与RGNNV聚类在一起,与SJNNV、BFNNV和TPNNV等其他鱼类神经坏死病毒亲缘关系较远,说明本研究分离得到的XMNNV属于RGNNV基因型。通过超速离心方法对病毒进行提纯,得到了纯化的NNV病毒。电镜观察结果表明,病毒粒子直径20 ~25 nm,结构为正二十面体,与已经报道的NNV结构一致。通过对XMNNV与其它RGNNV RNA2基因进行序列比对,在保守区设计引物,运用R T - P C R方法建立了 RGNNV的PCR检测方法,该方法灵敏度高达67 copies/uL 。纯化的病毒对E11 (条纹月鳢细胞系)和大黄鱼肌肉细胞进行感染,结果表明该病毒可以感染这两种鱼类细胞。E11细胞被感染病毒后,细胞出现空泡化,并最终导致细胞分解死亡;大黄鱼肌肉细胞感染后,细胞变圆,慢慢从培养皿壁脱落,最终解体死亡。另外,对感染后细胞进行P C R检测,结果显示为阳性,进一步确定了分离的NNV具有感染这两种细胞的能力。本研究通过电镜观察和PCR检测两种方法确定了患病石斑鱼携带NNV,通过对两种鱼类细胞的感染实验,确定了该病毒具有一定的感染能力。综上,本研究为石斑鱼NNV疾病的诊断提供了有效的方法,对石斑鱼NNV疾病的预防具有一定的指导意义。
闽南地区斜带石斑鱼(Epinephelus coioides)神经坏死病毒的基因型与分子进化研究
[J].本研究以闽南地区具有典型神经坏死病症状的斜带石斑鱼为材料,采用RT-PCR法对其进行病毒检测,对检测到的阳性序列进行双向测序和构建系统发育树,对病毒颗粒进行分离纯化,并通过分子进化模型分析病毒衣壳蛋白基因所受的选择压力。结果显示,采集到的6个石斑鱼样品均呈NNV 阳性,系统树分析发现6个样品的PCR扩增片段均为RGNNV基因型序列,表明闽南地区感染石斑鱼的神经坏死病毒主要为RGNNV基因型病毒;通过PEG法对病毒进行分离提純,获得直径为25~28 nm、呈二十面立体对称结构的无囊膜病毒 颗粒;分子进化分析显示NNV外壳蛋白基因经历了纯化选择,表明病毒在进化过程中没有 出现遗传变异并以相对恒定保守的速率进化。
Marker-assisted breeding of a lymphocystis disease-resistant Japanese flounder(Paralichthys olivaceus)
[J].
Prediction of total genetic value using genome-wide dense marker maps
[J].Recent advances in molecular genetic techniques will make dense marker maps available and genotyping many individuals for these markers feasible. Here we attempted to estimate the effects of approximately 50,000 marker haplotypes simultaneously from a limited number of phenotypic records. A genome of 1000 cM was simulated with a marker spacing of 1 cM. The markers surrounding every 1-cM region were combined into marker haplotypes. Due to finite population size N(e) = 100, the marker haplotypes were in linkage disequilibrium with the QTL located between the markers. Using least squares, all haplotype effects could not be estimated simultaneously. When only the biggest effects were included, they were overestimated and the accuracy of predicting genetic values of the offspring of the recorded animals was only 0.32. Best linear unbiased prediction of haplotype effects assumed equal variances associated to each 1-cM chromosomal segment, which yielded an accuracy of 0.73, although this assumption was far from true. Bayesian methods that assumed a prior distribution of the variance associated with each chromosome segment increased this accuracy to 0.85, even when the prior was not correct. It was concluded that selection on genetic values predicted from markers could substantially increase the rate of genetic gain in animals and plants, especially if combined with reproductive techniques to shorten the generation interval.
Genomic selection using extreme phenotypes and pre-selection of SNPs in large yellow croaker(Larimichthys crocea)
[J].
Predictive ability of genomic selection models for breeding value estimation on growth traits of Pacific white shrimp Litopenaeus vannamei
[J].
Accuracy of genomic evaluations of juvenile growth rate in common carp(Cyprinus carpio) using genotyping by sequencing
[J].Cyprinids are the most important group of farmed fish globally in terms of production volume, with common carp () being one of the most valuable species of the group. The use of modern selective breeding methods in carp is at a formative stage, implying a large scope for genetic improvement of key production traits. In the current study, a population of 1,425 carp juveniles, originating from a partial factorial cross between 40 sires and 20 dams, was used for investigating the potential of genomic selection (GS) for juvenile growth, an exemplar polygenic production trait. RAD sequencing was used to identify and genotype SNP markers for subsequent parentage assignment, construction of a medium density genetic map (12,311 SNPs), genome-wide association study (GWAS), and testing of GS. A moderate heritability was estimated for body length of carp at 120 days (as a proxy of juvenile growth) of 0.33 (s.e. 0.05). No genome-wide significant QTL was identified using a single marker GWAS approach. Genomic prediction of breeding values outperformed pedigree-based prediction, resulting in 18% improvement in prediction accuracy. The impact of reduced SNP densities on prediction accuracy was tested by varying minor allele frequency (MAF) thresholds, with no drop in prediction accuracy until the MAF threshold is set <0.3 (2,744 SNPs). These results point to the potential for GS to improve economically important traits in common carp breeding programs.
Predicting growth traits with genomic selection methods in zhikong scallop(Chlamys farreri)
[J].
Evaluation of genomic selection for seven economic traits in yellow drum(Nibea albiflora)
[J].
Genome-wide association improves genomic selection for ammonia tolerance in the orange-spotted grouper(Epinephelus coioides)
[J].
Genomic prediction of host resistance to sea lice in farmed Atlantic salmon populations
[J].
Genomic selection using BayesCπ and GBLUP for resistance against Edwardsiella tarda in Japanese flounder(Paralichthys olivaceus)
[J].
Genome-wide association and genomic prediction of resistance to viral nervous necrosis in European sea bass(Dicentrarchus labrax) using RAD sequencing
[J].European sea bass (Dicentrarchus labrax) is one of the most important species for European aquaculture. Viral nervous necrosis (VNN), commonly caused by the redspotted grouper nervous necrosis virus (RGNNV), can result in high levels of morbidity and mortality, mainly during the larval and juvenile stages of cultured sea bass. In the absence of efficient therapeutic treatments, selective breeding for host resistance offers a promising strategy to control this disease. Our study aimed at investigating genetic resistance to VNN and genomic-based approaches to improve disease resistance by selective breeding. A population of 1538 sea bass juveniles from a factorial cross between 48 sires and 17 dams was challenged with RGNNV with mortalities and survivors being recorded and sampled for genotyping by the RAD sequencing approach.We used genome-wide genotype data from 9195 single nucleotide polymorphisms (SNPs) for downstream analysis. Estimates of heritability of survival on the underlying scale for the pedigree and genomic relationship matrices were 0.27 (HPD interval 95%: 0.14-0.40) and 0.43 (0.29-0.57), respectively. Classical genome-wide association analysis detected genome-wide significant quantitative trait loci (QTL) for resistance to VNN on chromosomes (unassigned scaffolds in the case of 'chromosome' 25) 3, 20 and 25 (P < 1e06). Weighted genomic best linear unbiased predictor provided additional support for the QTL on chromosome 3 and suggested that it explained 4% of the additive genetic variation. Genomic prediction approaches were tested to investigate the potential of using genome-wide SNP data to estimate breeding values for resistance to VNN and showed that genomic prediction resulted in a 13% increase in successful classification of resistant and susceptible animals compared to pedigree-based methods, with Bayes A and Bayes B giving the highest predictive ability.Genome-wide significant QTL were identified but each with relatively small effects on the trait. Tests of genomic prediction suggested that incorporating genome-wide SNP data is likely to result in higher accuracy of estimated breeding values for resistance to VNN. RAD sequencing is an effective method for generating such genome-wide SNPs, and our findings highlight the potential of genomic selection to breed farmed European sea bass with improved resistance to VNN.
De novo assembly of a chromosome‐level reference genome of red-spotted grouper (Epinephelus akaara) using nanopore sequencing and Hi-C
[J].
Identification of candidate SNPs and genes associated with anti-RGNNV using GWAS in the red-spotted grouper,Epinephelus akaara
[J].
Fastp:an ultra-fast all-in-one FASTQ preprocessor
[J].
MultiQC:summarize analysis results for multiple tools and samples in a single report
[J].
Aligning sequence reads,clone sequences and assembly contigs with BWA-MEM
[J].
Sambamba:fast processing of NGS alignment formats
[J].Sambamba is a high-performance robust tool and library for working with SAM, BAM and CRAM sequence alignment files; the most common file formats for aligned next generation sequencing data. Sambamba is a faster alternative to samtools that exploits multi-core processing and dramatically reduces processing time. Sambamba is being adopted at sequencing centers, not only because of its speed, but also because of additional functionality, including coverage analysis and powerful filtering capability.Sambamba is free and open source software, available under a GPLv2 license. Sambamba can be downloaded and installed from http://www.open-bio.org/wiki/Sambamba.Sambamba v0.5.0 was released with doi:10.5281/zenodo.13200.© The Author 2015. Published by Oxford University Press.
The Genome Analysis Toolkit:A MapReduce framework for analyzing next-generation DNA sequencing data
[J].Next-generation DNA sequencing (NGS) projects, such as the 1000 Genomes Project, are already revolutionizing our understanding of genetic variation among individuals. However, the massive data sets generated by NGS--the 1000 Genome pilot alone includes nearly five terabases--make writing feature-rich, efficient, and robust analysis tools difficult for even computationally sophisticated individuals. Indeed, many professionals are limited in the scope and the ease with which they can answer scientific questions by the complexity of accessing and manipulating the data produced by these machines. Here, we discuss our Genome Analysis Toolkit (GATK), a structured programming framework designed to ease the development of efficient and robust analysis tools for next-generation DNA sequencers using the functional programming philosophy of MapReduce. The GATK provides a small but rich set of data access patterns that encompass the majority of analysis tool needs. Separating specific analysis calculations from common data management infrastructure enables us to optimize the GATK framework for correctness, stability, and CPU and memory efficiency and to enable distributed and shared memory parallelization. We highlight the capabilities of the GATK by describing the implementation and application of robust, scale-tolerant tools like coverage calculators and single nucleotide polymorphism (SNP) calling. We conclude that the GATK programming framework enables developers and analysts to quickly and easily write efficient and robust NGS tools, many of which have already been incorporated into large-scale sequencing projects like the 1000 Genomes Project and The Cancer Genome Atlas.
Twelve years of SAMtools and BCFtools
[J].
Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering
[J].Whole-genome association studies present many new statistical and computational challenges due to the large quantity of data obtained. One of these challenges is haplotype inference; methods for haplotype inference designed for small data sets from candidate-gene studies do not scale well to the large number of individuals genotyped in whole-genome association studies. We present a new method and software for inference of haplotype phase and missing data that can accurately phase data from whole-genome association studies, and we present the first comparison of haplotype-inference methods for real and simulated data sets with thousands of genotyped individuals. We find that our method outperforms existing methods in terms of both speed and accuracy for large data sets with thousands of individuals and densely spaced genetic markers, and we use our method to phase a real data set of 3,002 individuals genotyped for 490,032 markers in 3.1 days of computing time, with 99% of masked alleles imputed correctly. Our method is implemented in the Beagle software package, which is freely available.
PLINK:A tool set for whole-genome association and population-based linkage analyses
[J].Whole-genome association studies (WGAS) bring new computational, as well as analytic, challenges to researchers. Many existing genetic-analysis tools are not designed to handle such large data sets in a convenient manner and do not necessarily exploit the new opportunities that whole-genome data bring. To address these issues, we developed PLINK, an open-source C/C++ WGAS tool set. With PLINK, large data sets comprising hundreds of thousands of markers genotyped for thousands of individuals can be rapidly manipulated and analyzed in their entirety. As well as providing tools to make the basic analytic steps computationally efficient, PLINK also supports some novel approaches to whole-genome data that take advantage of whole-genome coverage. We introduce PLINK and describe the five main domains of function: data management, summary statistics, population stratification, association analysis, and identity-by-descent estimation. In particular, we focus on the estimation and use of identity-by-state and identity-by-descent information in the context of population-based whole-genome studies. This information can be used to detect and correct for population stratification and to identify extended chromosomal segments that are shared identical by descent between very distantly related individuals. Analysis of the patterns of segmental sharing has the potential to map disease loci that contain multiple rare variants in a population-based linkage analysis.
GCTA:A tool for genome-wide complex trait analysis
[J].
Efficient methods to compute genomic predictions
[J].Efficient methods for processing genomic data were developed to increase reliability of estimated breeding values and to estimate thousands of marker effects simultaneously. Algorithms were derived and computer programs tested with simulated data for 2,967 bulls and 50,000 markers distributed randomly across 30 chromosomes. Estimation of genomic inbreeding coefficients required accurate estimates of allele frequencies in the base population. Linear model predictions of breeding values were computed by 3 equivalent methods: 1) iteration for individual allele effects followed by summation across loci to obtain estimated breeding values, 2) selection index including a genomic relationship matrix, and 3) mixed model equations including the inverse of genomic relationships. A blend of first- and second-order Jacobi iteration using 2 separate relaxation factors converged well for allele frequencies and effects. Reliability of predicted net merit for young bulls was 63% compared with 32% using the traditional relationship matrix. Nonlinear predictions were also computed using iteration on data and nonlinear regression on marker deviations; an additional (about 3%) gain in reliability for young bulls increased average reliability to 66%. Computing times increased linearly with number of genotypes. Estimation of allele frequencies required 2 processor days, and genomic predictions required <1 d per trait, and traits were processed in parallel. Information from genotyping was equivalent to about 20 daughters with phenotypic records. Actual gains may differ because the simulation did not account for linkage disequilibrium in the base population or selection in subsequent generations.
Reliability of direct genomic values for animals with different relationships within and to the reference population
[J].Accuracy of genomic selection depends on the accuracy of prediction of single nucleotide polymorphism effects and the proportion of genetic variance explained by markers. Design of the reference population with respect to its family structure may influence the accuracy of genomic selection. The objective of this study was to investigate the effect of various relationship levels within the reference population and different level of relationship of evaluated animals to the reference population on the reliability of direct genomic breeding values (DGV). The DGV reliabilities, expressed as squared correlation between estimated and true breeding value, were calculated for evaluated animals at 3 heritability levels. To emulate a trait that is difficult or expensive to measure, such as methane emission, reference populations were kept small and consisted of females with own performance records. A population reflecting a dairy cattle population structure was simulated. Four chosen reference populations consisted of all females available in the first genotyped generation. They consisted of highly (HR), moderately (MR), or lowly (LR) related animals, by selecting paternal half-sib families of decreasing size, or consisted of randomly chosen animals (RND). Of those 4 reference populations, RND had the lowest average relationship. Three sets of evaluated animals were chosen from 3 consecutive generations of genotyped animals, starting from the same generation as the reference population. Reliabilities of DGV predictions were calculated deterministically using selection index theory. The randomly chosen reference population had the lowest average relationship within the reference population. Average reliabilities increased when average relationship within the reference population decreased and the highest average reliabilities were achieved for RND (e.g., from 0.53 in HR to 0.61 in RND for a heritability of 0.30). A higher relationship to the reference population resulted in higher reliability values. At the average squared relationship of evaluated animals to the reference population of 0.005, reliabilities were, on average, 0.49 (HR) and 0.63 (RND) for a heritability of 0.30; 0.20 (HR) and 0.27 (RND) for a heritability of 0.05; and 0.07 (HR) and 0.09 (RND) for a heritability of 0.01. Substantial decrease in the reliability was observed when the number of generations to the reference population increased [e.g., for heritability of 0.30, the decrease from evaluated set I (chosen from the same generation as the reference population) to II (one generation younger than the reference population) was 0.04 for HR, and 0.07 for RND]. In this study, the importance of the design of a reference population consisting of cows was shown and optimal designs of the reference population for genomic prediction were suggested.Copyright © 2012 American Dairy Science Association. Published by Elsevier Inc. All rights reserved.
The importance of information on relatives for the prediction of genomic breeding values and the implications for the makeup of reference data sets in livestock breeding schemes
[J].
Effects of marker density and population structure on the genomic prediction accuracy for growth trait in Pacific white shrimp Litopenaeus vannamei
[J].
Evaluation for the effect of low-coverage sequencing on genomic selection in large yellow croaker
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}